
JOURNAL OF CLINICAL PEDIATRICS RESEARCH
On this page
Arthrogryposis Renal Dysfunction Cholestasis Syndrome: An Uncommon Presentation in a Newborn
Paramesh Sreekumar1*, Nayla Ali Alfaw2, Minoosh Nasef3 and Emad Shatla4
1Registrar, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain
2Registrar, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain
3Consultant, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain
4Consultant, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain
*Corresponding Author: Paramesh Sreekumar, Registrar, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain.
| ReceivedFeb 1, 2024 | RevisedFeb 20, 2024 | AcceptedFeb 28, 2024 | PublishedMar 20, 2024 |
Abstract
A preterm baby born at 35 weeks of gestation was noted to have severe limb deformities suggestive of arthrogryposis along with dysmorphic facies and developed seizures, renal dysfunction in addition to cholestasis. MRI brain showed corpus callosum agenesis. Although there are several isolated cases of arthrogryposis, the association with other congenital anomalies are rarely seen. The constellation of anomalies noted in this case along with renal tubular dysfunction and direct hyperbilirubinemia with normal GGT is suggestive of Arthrogryposis renal dysfunction Cholestasis (ARC) syndrome. It is a very rare autosomal recessive multisystem disorder resulting from mutations in the vacuolar protein sorting 33B (VPS33B) or VPS33B-interacting protein, and apical-basolateral polarity regulator (VIPAR) genes. This disorder is associated with a very poor prognosis with children seldom surviving beyond 1 year of age necessitating a detailed genetic evaluation to identify the various mutations and appropriate genetic counselling.
Keywords
Preterm; Arthrogryposis; Cholestasis; Renal dysfunction
Introduction
Arthrogryposis multiplex congenital (AMC) in newborn babies are a group of genetic disorders characterized by the presence of multiple contractures with resultant limb deformities in at least
two or more joints, that occur prior to birth [1]. Most cases occur sporadically with around 400 gene mutations identified [2]. The prevalence is estimated to be 1 in 3000 live births [3].
A rare presentation known as Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome is a multisystem disorder which is inherited in an autosomal recessive manner and caused by mutations in the vacuolar protein sorting 33B (VPS33B) or VPS33B-interacting protein, and less commonly apical-basolateral polarity regulator (VIPAR) genes [4].
It is characterized by severe limb contractures, renal dysfunction, neonatal cholestasis with a low or normal low gamma-glutamyl transferase (GGT). There could be additional features like platelet abnormalities, corpus callosum agenesis, cardiovascular disease, ichthyosis, recurrent infections and hearing loss [5].
It was first recognized in two children born to parents of a consanguineous family in 1973 [6]. Several cases are reported from countries like Saudi Arabia and Pakistan, where there are high rates of consanguinity, but also few sporadic cases were noted from other parts of the world [7,8].
There is no definitive treatment available for this condition and it has a poor prognosis. Here, we present a preterm baby girl born with multiple congenital anomalies, in particular limb deformities suggestive of arthrogryposis, renal dysfunction and cholestasis which indicated toward Arthrogryposis Renal dysfunction Cholestasis (ARC) syndrome.
Case
A preterm baby girl was born at 35 weeks of gestation by emergency caesarean section due to fetal distress. The baby’s birth weight was 2 kg. Mother was 25 years old primigravida with no known medical illness and an antenatal period without any complications. Baby required resuscitation at birth with APGARs of 5, 8 at 1 minute and 5 minutes respectively.
The baby was noted to have dysmorphic features characterized by short neck, low set ears, frontal bossing, brachydactyly and severe flexion deformities of the lower limbs. There were no anomalies detected in the antenatal scans. The baby was born out of a non-consanguineous marriage and there was no family history of any congenital disorders. The baby had severe respiratory distress at birth requiring ventilation and surfactant therapy. The patient was commenced on fluids containing 10 percent dextrose and intravenous antibiotics.
Initially blood sugars were not maintained in the normal range but stabilized following an increase in the total fluid intake and change of fluid dextrose concentration to 12.5 percent. There was also severe hyponatremia (Na 118 mmol/l) noted on day 1 of life requiring correction with 3% sodium chloride. However, hyponatremia persisted with normal urine output and increased urinary excretion of sodium and chloride. The urine osmolarity was normal. Cardiac murmur was detected at birth for which an echocardiography was done that revealed a 2 mm PDA with bidirectional shunt, right sided dilatation and a pulmonary gradient of 50 mm Hg. The baby remained hemodynamically stable. And developed abnormal movements on day 7 of life which were controlled with anti-convulsant medication levetiracetam. MRI brain done on day 12 of life revealed corpus callosum agenesis (Figure 1). Ophthalmological evaluation was normal. Initial laboratory investigations showed normal haemoglobin levels and white blood cell count but low platelets. There were no abnormal cells and platelet morphology was normal in the peripheral smear. Liver function showed elevated direct fraction of bilirubin with normal liver enzymes including GGT. There was slight increase in the gallbladder wall thickness, normal liver echotexture in abdominal ultrasound and no evidence of any biliary obstruction. No renal anomalies were detected. Genetic studies were not done. The baby continued to remain on the ventilator, developed sepsis with disseminated intravascular coagulation and expired on day 22 of life.
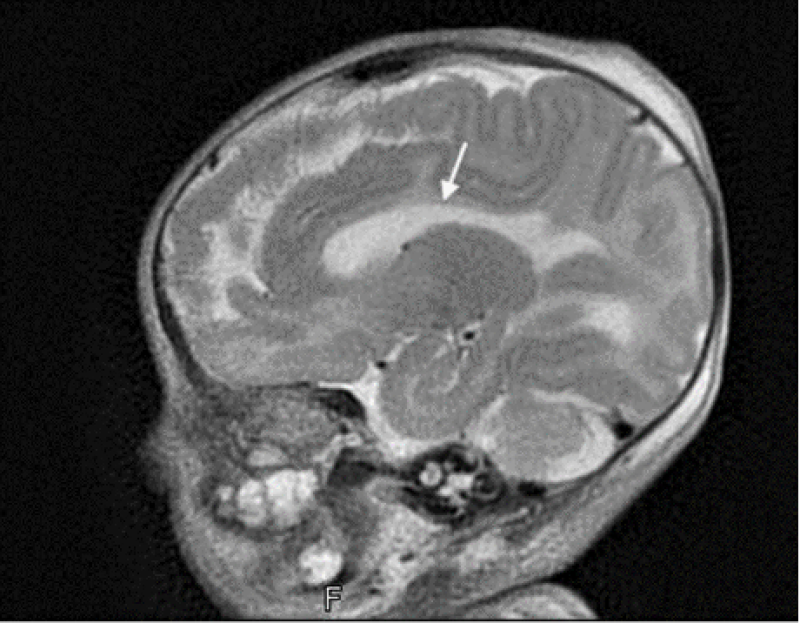
Figure 1: MRI Brain image suggestive of corpus callosum agenesis.
Discussion
In this case, the presence of multiple congenital anomalies in particular arthrogryposis of the limbs, renal dysfunction as evidenced by persistent hyponatremia secondary to increased urinary sodium loss and glycosuria along with direct hyperbilirubinemia with normal GGT levels were similar to the manifestations seen in Arthrogryposis Renal dysfunction (ARC) syndrome. Additional features associated with this syndrome like corpus callosum agenesis and platelet abnormalities were also seen in the patient. During embryonic development in utero, limitation of movement due to uterine malformations, multiple gestation, amniotic bands or oligohydramnios have been attributed to the occurrence of arthrogryposis. Genetic disorders like myopathies including muscular dystrophies, connective tissue disorders, anterior horn cell disease have been found to predispose to babies born with multiple limb contractures. Congenital infections with cytomegalovirus, rubella virus and more recently zika virus and maternal immune diseases such as myasthenia gravis are also found to be linked with these disorders.
Bamshad, et al. [9], categorized Arthrogryposis multiplex congenita (AMC) in 2009 into three major types: amyoplasia, distal arthrogryposis, and syndromic forms.
Amyoplasia or classic arthrogryposis were mainly sporadic cases and presents with multiple limb contractures and normal intelligence in the affected children. About 10% of patients have Gastrointestinal anomalies like gastroschisis, bowel atresia.
Distal arthrogryposis involves the hands and feet, but the large joints are spared. Many distal arthrogryposes are transmitted as autosomal dominant disorders, but X-linked mutations are known. A syndromic form of AMC is suspected when there is a constellation of other congenital anomalies along with limb contractures like renal dysfunction and cholestasis and points towards ARC syndrome. The mechanisms involved are unclear, but considered due to VPS33B gene mutations resulting in abnormal expression of VPS33B protein which is involved in establishing structural and functional aspects of hepatocyte polarity. VPS33B and VIPAR proteins form a complex that is responsible for maintaining apical-basolateral polarity as well as normal cellular structure [10].
The main triad of clinical presentation of ARC includes congenital joint contractures, renal tubular dysfunction, and cholestasis. Less common features include central nervous system malformation like agenesis of corpus callosum, platelet anomalies, ichthyosis, failure to thrive, congenital cardiovascular anomalies, deafness, recurrent infection, and bleeding due to coagulation dysfunction [5,11].
Renal tubular dysfunction may manifest as renal tubular acidosis, Fanconi syndrome, which was noted in the case. There is an association noted with nephrogenic diabetes insipidus [12] contrary to the case where babies were presented with fluid retention and persistent hyponatremia.
There was a case of ARC syndrome reported recently with absent kidney [13].
Another cardinal feature of this syndrome is the presence of cholestasis with low γ-glutamyl transpeptidase (GGT) levels signifying no biliary obstruction with mildly elevated alanine aminotransferase (ALT)/aspartate aminotransferase (AST), and liver biopsy showing histological features of giant cell transformation of hepatocytes, bile duct paucity, and lipofuscin deposition [11].
There have been rare instances of cases with cholestasis and high GGT [14].
Other less common manifestation of platelet abnormalities thrombocytopenia was seen in the patient similar to another reported case [15], although the platelet morphology was normal in the peripheral smear of this case. There was another case by Gupta V, et al. [13], that showed no abnormality in platelet morphology. Deal, et al. [16], found large platelets which lacked alpha granules and had low concentrations of thromboglobulin, platelet factor 4 and thrombospondin. Reports of VPS33B gene mutations leading to a similar disease, known as autosomal recessive keratoderma-ichthyosis-deafness syndrome (ARKID) have been noted [17].
The typical features of ARKID include ichthyosis, osteopenia, hearing loss and severe failure to thrive. A recent study was reported which described patients with a mild phenotype of ARC syndrome with VPS33B gene variants and prolonged survival [18]. Patients with the c.1225+5G>C mutation in the VPS33B gene may have a mild phenotype and a longer lifespan [19].
The diagnosis of this condition is mainly based on clinical presentation and sequencing analysis for VPS33B and VIPAR gene mutations. The analysis of VPS33B protein expression in skin fibroblasts and platelet morphology in peripheral blood smears are newer alternative techniques which can aid in the diagnosis [11]. Treatment is mainly supportive, consisting of ursodeoxycholic acid and fat-soluble vitamin administration, maintenance of acid–base and electrolyte balance. Surgical correction for joint contracture can restore some mobility. The management of these cases is often difficult owing to the multisystem involvement and poor prognosis. In these patients, sepsis, bleeding, severe dehydration and acidosis are the main causes of morbidity and mortality. Treatment modalities are mainly supportive in nature and often do not yield satisfactory results. The use of ursodeoxycholic acid and biliary diversion are of some benefit in cholestasis cases [20]. Bleeding episodes can be controlled by the use of prophylactic platelet transfusions and ε-aminocaproic acid in some patients [21]. A report showed the beneficial role of liver transplantation to treat cholestasis [22]. The prognosis of ARC syndrome is often poor and most of the patients do not survive beyond the first year of life [23].
Conclusion
ARC syndrome is a rare multisystem disorder, which is often detected based on the classical clinical presentation of arthrogryposis, renal dysfunction and neonatal cholestasis. It requires further analysis of the associated gene mutations, notably that of VPS33B gene. There is no known specific treatment and this condition harbours a poor prognosis. Therefore, a detailed analysis of genetic mutation facilitates accurate diagnosis and helps in providing appropriate genetic counselling to the family. Further studies are needed in the field of medical genetics to identify various phenotypic variants and for a possible breakthrough gene therapy as a curative option.
References
1. Cachecho S, Elfassy C, Hamdy R, Rosenbaum P, Dahan‐Oliel N. Arthrogryposis Multiplex Congenita Definition: Update Using An International Consensus‐based Approach. Am J Med Genet C Semin Med Genet. 2019;181(3):280-287. PubMed | CrossRef
2. Kiefer J, Hall JG. Gene Ontology Analysis of Arthrogryposis (Multiple Congenital Contractures). Am J Med Genet C Semin Med Genet. 2019;181(3):310-326. PubMed | CrossRef
3. Wahlig B, Poppino K, Jo CH, Rathjen K. Arthrogryposis Multiplex Congenita: A 28-year Retrospective Study. Dev Med Child Neurol. 2022;64(4):476-80. PubMed | CrossRef
4. Cullinane AR, Straatman‐Iwanowska A, Seo JK, Ko JS, Song KS, Gizewska M, et al. Molecular Investigations to Improve Diagnostic Accuracy in Patients with ARC Syndrome. Hum Mutat. 2009;30(2):E330-7. PubMed | CrossRef
5. Agawu A, Sheppard S, Lin HC. A novel VPS33B Mutation Causing a Mild Phenotype of Arthrogryposis, Renal Dysfunction, and Cholestasis Syndrome. J Pediatr Gastroenterol Nutr. 2019;69(2):e55-6. PubMed | CrossRef
6. AR LR. Familiare Gallengansmissbildungen Mit Tubular Neireninsurfizienz. Helv Paediatr Acta. 1973;28:1-2.
7. Abu-Sa'da O, Barbar M, Al-Harbi N, Taha D. Arthrogryposis, Renal Tubular Acidosis and Cholestasis (ARC) Syndrome: Two New Cases and Review. Clin Dysmorphol. 2005;14(4):191-6. PubMed | CrossRef
8. Tekin N, Durmuş-Aydoğdu S, Dinleyici EC, Bör O, Bildirici K, Akşit A. Clinical and Pathological Aspects of ARC (Arthrogryposis, Renal Dysfunction and Cholestasis) Syndrome in two Siblings. Turk J Pediatr. 2005;47(1):67-70. PubMed
9. Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: A Review and Update. J Bone Joint Surg Am. 2009;91(Suppl 4):40-6. PubMed | CrossRef
10. Hanley J, Dhar DK, Mazzacuva F, Fiadeiro R, Burden JJ, Lyne AM, et al. Vps33b is Crucial for Structural and Functional Hepatocyte Polarity. J Hepatol. 2017;66(5):1001-11. PubMed | CrossRef
11. Zhou Y, Zhang J. Arthrogryposis–renal Dysfunction-cholestasis (ARC) Syndrome: From Molecular Genetics to Clinical Features. Ital J Pediatr. 2014;40(1):1-7. PubMed | CrossRef
12. Malaki M, Mandana R, Ghaffari S. ARC Syndrome with Complex Renal Problems: Nephrocalcinosis, Proximal and Hyperkalemic Distal RTA and Nephrogenic Diabetes Insipidus. Saudi J Kidney Dis Transpl. 2012;23(4):804-9. PubMed | CrossRef
13. Gupta V, Pandita A, Panghal A, Kallem V. Arthrogryposis, Renal Dysfunction and Cholestasis (ARC) Syndrome: A Rare Association with High GGT Level and Absent Kidney. BMJ Case Rep. 2018;2018:bcr-2017. PubMed | CrossRef
14. Wang JS, Zhao J, Li LT. ARC Syndrome with High GGT Cholestasis Caused by VPS33B Mutations. World J Gastroenterol. 2014;20(16):4830. PubMed | CrossRef
15. Saadah OI, Bokhari BE, Alshaeri TM, Jastaniah W. Haematological Manifestations of Arthrogryposis-renal Dysfunction-cholestasis (ARC) Syndrome: A Case Report. Arab J Gastroenterol. 2013;14(1):26-8. PubMed | CrossRef
16. Deal JE, Barratt TM, Dillon MJ. Fanconi Syndrome, Ichthyosis, Dysmorphism, Jaundice and Diarrhoea: A New Syndrome. Pediatr Nephrol. 1990;4:308-13. PubMed | CrossRef
17. Gruber R, Rogerson C, Windpassinger C, Banushi B, Straatman-Iwanowska A, Hanley J, et al. Autosomal Recessive Keratoderma-ichthyosis-deafness (ARKID) Syndrome is Caused by VPS33B mutations Affecting Rab Protein Interaction and Collagen Modification. J Invest Dermatol. 2017;137(4):845-54. PubMed | CrossRef
18. Linhares ND, Fagundes ED, Ferreira AR, Queiroz TC, da Silva LR, Pena SD. Mild Phenotype of Arthrogryposis, Renal Dysfunction, and Cholestasis Syndrome 1 Caused by a Novel VPS33B Variant. Front Genet. 2022;13:796759. PubMed | CrossRef
19. Duong MD, Rose CM, Reidy KJ, Del Rio M. An Uncommon Case of Arthrogryposis, Renal Dysfunction, and Cholestasis (ARC) Syndrome and Review of the Renal Involvement: Answers. Pediatr Nephrol. 2020;35:249-51. PubMed | CrossRef
20. Smith H, Galmes R, Gogolina E, Straatman‐Iwanowska A, Reay K, Banushi B, et al. Associations among Genotype, Clinical Phenotype, and Intracellular Localization of Trafficking Proteins in ARC Syndrome. Hum Mutat. 2012;33(12):1656-64. PubMed | CrossRef
21. Weyand AC, Lombel RM, Pipe SW, Shavit JA. The Role of Platelets and ε‐Aminocaproic Acid in Arthrogryposis, Renal Dysfunction, and Cholestasis (ARC) Syndrome Associated Hemorrhage. Pediatr Blood Cancer. 2016;63(3):561-3. PubMed | CrossRef
22. Dehghani SM, Bahador A, Nikeghbalian S, Salahi H, Geramizadeh B, Malekpour A, et al. Liver Transplant in a Case of Arthrogryposis-renal Tubular Dysfunction-cholestasis Syndrome with Severe Intractable Pruritus. Exp Clin Transplant. 2013;11(3):290-2. PubMed | CrossRef
23. Gissen P, Tee L, Johnson CA, Genin E, Caliebe A, Chitayat D, et al. Clinical and Molecular Genetic Features of ARC Syndrome. Hum Genet. 2006;120:396-409. PubMed | CrossRef
Paramesh Sreekumar1*, Nayla Ali Alfaw2, Minoosh Nasef3 and Emad Shatla4
1Registrar, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain
2Registrar, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain
3Consultant, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain
4Consultant, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain
*Corresponding Author: Paramesh Sreekumar, Registrar, Department of Pediatrics and Neonatology, King Hamad University Hospital, Kingdom of Bahrain.
Copyright© 2024 by Sreekumar P, et al. All rights reserved. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Sreekumar P, Alfaw NA, Nasef M, Shatla E. Arthrogryposis Renal Dysfunction Cholestasis Syndrome: An Uncommon Presentation in a Newborn. J Clin Ped Res. 2024;3(1):57-63. DOI: https://doi.org/10.37191/Mapsci-2583-4525-3(1)-018